Research
We are interested in developing an understanding, at the molecular and atomic level, of basic biochemical and biological processes. The long-range goal of our research is to study the interactions between pathogens and hosts, in order to describe mechanisms of pathogen engagement of target cells and to provide a basis for vaccine and drug design. We study viruses as well as bacterial pathogens. We use protein crystallography, in combination with associated biochemical techniques (site-directed mutagenesis, surface plasmon resonance, circular dichroism spectroscopy, titration calorimetry etc), to establish the three-dimensional structure and function of functionally relevant proteins alone and in complex with ligands.
A particular emphasis is placed on understanding basic principles of protein-protein and protein-carbohydrate interactions. Many such interactions are well characterized at a functional level, yet poorly understood at the molecular or atomic level. However, such knowledge is needed to develop compounds that are able to modulate or block biological interactions. For example, by studying the determinants of viral attachment to host cells and establishing principles of attachment, one can develop molecules that interfere with binding. It also becomes possible to redirect or retarget viruses to different receptors, which is highly relevant for gene delivery approaches.
We are interested in the following problems:
Virus-Host Interactions
- Measles virus and its receptors, CD46 and SLAM
- Human adenovirus-host cell interactions
- Attachment of human reovirus to its receptors Junctional Adhesion Molecule-A and sialic acid
- Engagement of ganglioside receptors by polyomaviruses (DNA tumor viruses)
Staphylococcal proteins as potential drug targets
- Structure and function of enzymes involved in the bacterial cell wall synthesis and degradation
- Stucture and function of enzymes involved in staphylococcus biofilm production
- Stuctural genomics: Virulence factors of Staphylococcus aureus
Other bacterial pathogens
- Adenylyl cyclase from Mycobacterium tuberculosis
Structure and function of human NK cell receptors
- Structural analysis of the NKC encoded, C-type lectin like receptor-ligand pair Nkp80 and AICL
Structural basis of ligand-mediated signalling in eukaryotes and plants
- Deciphering the structure of Insulin Receptor Substrate
Modern biochemical and biomedical research addresses complex problems. This often necessitates worldwide collaborations and multiple research teams that work together. Our group participates in a large number of international collaborations, and we also part of several multi-investigator research programs. Our projects are supported by the following national and international scientific networks, as well as by industry.
Virus-host interactions
Measles virus and its receptors, CD46 and SLAM
Paramyxoviruses such as measles virus, respiratory syncytial virus and the parainfluenza viruses are major causes of morbidity and mortality worldwide. Measles virus has two receptors. The first, human CD46, is a cell-surface glycoprotein whose normal function is to regulate complement and prevent autologous tissue from complement-mediated destruction. The second, Signalling Lymphocytic Activation Molecule (SLAM) is a T-cell costimulatory protein that plays a prominent role in the immune response of the host. Understanding the precise interactions between measles virus and its receptors is critical for the design of effective antiviral compounds. We have already established the crystal structure of the CD46 measles-virus binding fragment (PubMed), and we are now working on determining the structures of the complete CD46 ectodomain and of SLAM. Additional efforts are on probing the virus-receptor interaction through site-directed mutagenesis.
Human adenovirus-host cell interactions
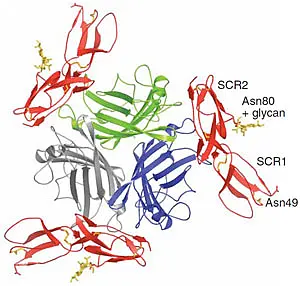
The measles virus receptor, CD46, has also been identified as the receptor for a class of human herpesviruses and for group B adenoviruses. Adenoviruses (Ads) have a non-enveloped icosahedral capsid with a linear double-stranded DNA. The major capsid proteins are the hexon, the penton base and the fiber. We have solved several structures of the species B adenovirus attachment protein, fiber knob (Ad7, Ad11, Ad14 and Ad21), as well as two complex structures of the Ad11 and Ad21 fiber knobs with the binding fragment of the cellular receptor CD46 (PubMed, PubMed). The figure on the left shows the structure of the Ad11 fiber knob (green, blue, grey) in complex with CD46 (red). Our previous results provide a profound insight into the variability of CD46 binding among species B adenoviruses. The different binding mode correlates with different binding affinity and likely also with tissue tropism. Our aim is to further investigate the attachment processes of adenoviruses via X-ray crystallography.
Furthermore, our objective is to elucidate interactions between other adenovirus capsid proteins like the penton base and the hexon, with their respective binding partners, integrin receptors and blood coagulation factor X, on a molecular level. The structure of Ad2 penton base has been solved via X-ray crystallography. Yet, the penton base binding region, a protruding loop containing an RGD motif, could not be traced. The RGD binding motif is shared by several other viruses like the foot-and-mouth-disease virus, the human herpesviruses 1 and 8, Hantaviruses and Rotaviruses. Thus, elucidation of the interactions between the penton base and integrin receptors may provide insight into the internalization process of several viruses, and constitute a platform for drug development against infections by different viruses that share a common step of infection. Moreover, the interaction between penton base and integrins is a promising target for gene therapy, as adenovirus is capable of escaping antibody neutralization by antibodies directed to the RGD binding site. Thus, analysis of virus-receptor interactions on a molecular level will help to determine factors that define the pathogenicity and gene delivery properties of adenoviruses and may help to develop antiviral drugs, as well as gene delivery applications. This project is funded by the Deutsche Forschungsgemeinschaft / Sonderforschungsbereich 685.
Attachment of human reovirus to its receptors Junctional Adhesion Molecule-A and sialic acid
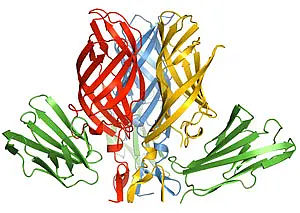
The mammalian reoviruses are important models for studies of viral pathogenesis and replication, and in particular for analysis of viral determinants of central nervous system injury. They are oncolytic viruses, lysing tumor cells specifically, and are therefore used in clinical trials in cancer therapy. The reovirus attachment protein sigma1 plays a central role in receptor binding specificity and in determining the pathway of viral spread. Sigma1, a fiber-like protein that has an elongated tail and a globular head, is located at the 12 vertices of the icosahedral virion. It has binding sites for JAM-A (junctional adhesion molecule-A) within the head and for and sialic acid within the tail. Our crystal structure of a JAM-binding fragment of sigma1 reveals an elongated trimer with two domains: a compact head with a beta-barrel fold and a fibrous tail containing a triple beta-spiral. Numerous structural and functional similarities between reovirus sigma1 and the adenovirus fiber suggest a totally unexpected evolutionary link in the receptor-binding strategies of these two viruses.
The fibrous tail is mainly responsible for sigma1 trimer formation, and it contains a highly flexible region that allows for significant movement between the base of the tail and the head. The architecture of the trimer interface and the observed flexibility indicate that sigma1 is a metastable structure poised to undergo conformational changes upon viral attachment and cell entry. We have since crystallized the receptor for sigma1, JAM-A (PubMed), and have also characterized the interaction with the virus biochemically (PubMed) and solved the structure of the complex between sigma1 and JAM-A (PubMed). The complex is shown in the figure on the left, with sigma1 shown in red, blue, and yellow, and JAM-A in green. We are presently engaged in crystallizing sigma1 of other reovirus serotypes in complex with JAM-A and carbohydrates, and in probing the determinants of the conformational changes of sigma1 that we have identified. Studying the sigma1 proteins from different reovirus strains will help in understanding how sequence differences translate into the known different behavior of these strains in the host. Our studies are done in collaboration with Terence Dermody at the Vanderbilt University School of Medicine in Nashville, Tennessee. This project is funded by the National Institutes of Health.
Engagement of ganglioside receptors by polyomaviruses (DNA tumor viruses)

Viruses must attach to specific receptors on their host cells in order to initiate entry, but too tight receptor binding prevents viral progeny from spreading to new host cells. As a result, attachment and release processes depend on precisely regulated contacts and affinities between viral proteins and their cognate ligands at the cell surface.
Polyomaviruses are a group of small, non-enveloped DNA viruses that can infect birds, rodents and primates. Many of them can cause tumors or transform cells in culture. Polyomaviruses that infect humans cause asymptomatic persistent infections in healthy individuals. However, BK virus and JC virus can be reactivated in patients with damaged immune systems. They lead to graft loss in transplant recipients or a fatal demyelinating disease in AIDS patients, respectively. The recently identified Merkel cell polyomavirus is the likely cause of the rare but highly aggressive Merkel cell tumor of the human skin.
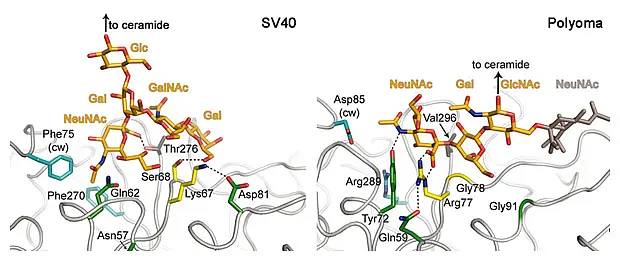
Most polyomaviruses attach to gangliosides on host cells via their major capsid protein VP1. On the figure on the left, you can see a model of SV40 attaching to gangliosides on the cell surface. Gangliosides are complex glycolipids residing primarily in lipid rafts and are usurped by polyomaviruses for transport to the endoplasmic reticulum, a crucial step during viral infection. Therefore, receptor binding is an interesting target for antiviral drugs. Despite the high homology of VP1 proteins, even closely related viruses bind different gangliosides. This directly determines their species and tissue tropism, pathogenicity and spread, but the molecular interactions at the basis of this behaviorare not known.
We are working to determine the structures of different VP1 proteins together with their carbohydrate receptors. For several viruses, the carbohydrate ligands are not known and we use glycan array screening to define their receptor specificity. In addition, different affinities for several receptors contribute e.g. to viral spread, so we also determine the binding affinity for the VP1 carbohydrate interaction. Taken together, this data will provide a detailed view how structural changes alter the receptor specificity of these viruses and thus alter their pathogenicity and host range. We have previously done such studies for the model organisms murine polyomavirus (PubMed) and simian virus 40 (PubMed) (see figure below) and are now expanding the project to human pathogens and their close relatives.
We do this project in collaboration with Walter Atwood (Brown University), Robert Garcea (University of Colorado, Boulder), Dale Mierke (Dartmouth College), the Consortium for Functional Glycomics Core H (Emory University) and Ten Feizi (Imperial College). It is funded by the German Research Foundation (SFB 685) and an NIH program project grant with Drs. Atwood and Mierke.
Staphylococcal proteins as potential drug targets
Structure and function of enzymes involved in the bacterial cell wall synthesis and degradation
PknB. Staphylococcus aureus (S. aureus) is a gram-positive bacterium colonising mainly the upper respiratory tract and the skin. The pathogen causes disease patterns ranging from mild skin or respiratory infections to life-threatening diseases such as endocarditis, sepsis or toxic shock syndrome. These life-threatening infections occur mostly in persons with multiple risk factors for infections. The increasing amount of multi-drug resistant strains has become a severe problem in the treatment of these bacterial infections. Therefore, new approaches for therapy are urgently needed due to mounting resistance against the existing drugs that target S. aureus. Protein kinases are important enzymes for regulation of processes and signal transduction pathways in prokaryotes and eukaryotes. While many microorganisms encode several serine/threonine protein kinases (STPKs), S. aureus encodes only one such protein, PknB. Thus, PknB provides a promising target in the treatment of S. aureus infections. We therefore attempt to solve the X-ray crystallographic structure of PknB at high resolution.
MprF. Defensins are positively charged antimicrobial peptides of the innate immune system that protect humans against microbial infections. MprF is an integral membrane protein rendering S. aureus insensitive against these defensins. A synthethase activity in MprF adds l-lysin to phosphatidylglycerol. The resulting l-lysin-phosphatidylglycerol is built into the bacterial membrane. This leads to a change in the net surface charge of the bacterium, which in turn prevents the cationic defensins from attacking the bacterium. This makes MprF a possible drug target to defeat S. aureus. We are working on the purification of the MprF synthetase domain in order to crystallize it and solve its X-ray structure.
Amidase. The major autolysins (Atl) of S. epidermidis and S. aureus play an important role in cell separation, and their mutants are also attenuated in virulence. Therefore, autolysins represent a promising target for the development of new types of antibiotics. We attempt to solve the resolution structure of the catalytically active amidase domain AmiE (amidase S. epidermidis) from the major autolysin of S. epidermidis. We are also trying to identify the minimal peptidoglycan fragment that can be used as a substrate by the enzyme using molecular docking and digestion assays. The results will provide an excellent platform for the design of specific inhibitors that target staphylococcal cell separation and can thereby prevent growth of this pathogen.
Structure and function of enzymes involved in staphylococcus biofilm production
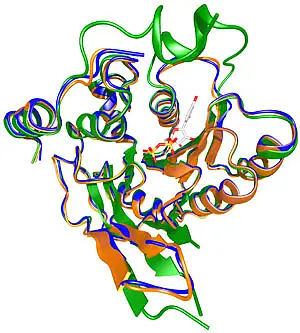
A basic model mechanism has been formulated for staphylococcal growth on artificial polymer surfaces, involving initial adhesion and subsequent accumulation, but mechanistic details are still unclear. The research on biofilm production is of clinical interest, as patients with implanted medical devices, e.g., polyethylene prostheses and silastic catheters generally coated with a proteinaceous conditioning film, do suffer severe nosocomial sepsis due to chronic infections by pathogenic coagulase-negative agents such as S. epidermidis. While the adhesion is brought on by several cell surface proteins and PS/A (polysaccharide/adhesin), the accumulation process of the species is mediated by a biofilm excess. Biofilm production itself is a very common phenotype of S. epidermidis clinical isolates among other genera. It requires a regulated membrane-situated enzyme apparatus which turns on when the bacterial cells start growing. The current work is aimed at solving the molecular structure of IcaA (functional homologue of SpsA from Bacillus subtilis), the enzymatic component of the protein complex. On the left, the SpsA crystal structure is shown (green), together with the IcaA homology models for S. epidermidis (orange) and S. aureus (blue). A further aim to help in developing therapeutics by evaluating potential inhibitors in protein interaction assays for fast-spreading multi-resistant specimen in clinical environment. Funding is derived from the SFB766.
Structural genomics: Virulence factors of Staphylococcus aureus
S. aureus is a gram-positive bacterium that colonizes the anterior nares of at least one third of the human population. It is also known to cause a variety of infections, ranging from superficial lesions to severe systemic infections. The pathogenicity of this organism largely depends on the successful adaptation to the human host and the environmentally coordinated expression of virulence factors. A network of interacting regulons tightly controls the virulence factor expression pattern during the growth cycle of S. aureus.
The agr (accessory gene regulator), the SarA (staphylococcal accessory regulator), the Sigma (alternative sigma factor) and the Rot (regulator of toxins) regulons are among the best- characterized virulence-associated regulons. More recently, the sae locus was described as a regulatory locus consisting of four open reading frames, two of which encode the response regulator SaeR and the sensor histidin kinase SaeS, respectively. The sae operon gives rise to the two-component system SaeRS, which is now known as another crucial regulator of virulence gene expression in S. aureus. Expression of a multitude of virulence factors, such as serine protease SspA, thermonuclease Nuc, coagulase Coa, alpha-hemolysin Hla, beta-hemolysin Hlb and fibronectin binding protein FnbA, was shown to be activated by the SaeRS system. It has also been proposed that SaeRS regulates the synthesis of extracellular proteins downstream of the agr regulon and might modify quorum sensing-dependent regulation. More recently, proteomic and transcriptomic studies were carried out in the S. aureus, yielding a the complete set of proteins that are most likely under the control of SaeRS. Most of these proteins play a role in immune evasion, while others are especially involved in adhesion to host cells. Although the detailed functionality of these virulence factors is not known to date, we will assign a function by structural genomics. Therefore our research is designed to determine the structure of these proteins and then to deduce a biological function by comparison to structurally homologous proteins. This work is funded by SFB/Transregio 34.
Other bacterial pathogens
Adenylyl cyclase from Mycobacterium tuberculosis
Mycobacterium tuberculosis is one of the most effective pathogens. It claims about 2 million lives each year and lurks in one-third of the world’s population. It is possible that M. tuberculosis has evolved eukaryotic-like signal transduction mechanisms which may be capable of modulating host cell signal pathways. In M. tuberculosis, 15 genes encode class III adenylyl cyclases of variant domain compositions, suggesting that cAMP formation may contribute to the pathogenesis.
We are interested in the structure, function and regulation of the mammalian-like adenylyl cyclase Rv1625c from M. tuberculosis. Adenylyl cyclase catalyzes the formation of cAMP which is a key signaling molecule in virtually all living organisms. Up to now, the most intensively studied adenylyl cyclases are nine mammalian membrane-bound enzymes. They contain two putative membrane domains; each with six transmembrane spans, and two complementary catalytic domains. The structure of the catalytic domains is well characterized; they form a pseudoheterodimer. The membrane domains account for about 40% of the protein, the function of which is unknown. Hitherto, no structure details are available for the holoenzymes because of the scarcity of the proteins.
Rv1625c from M. tuberculosis is a mammalian-like adenylyl cyclase with six putative transmembrane helices and a catalytic domain, corresponding exactly to one half of the nine mammalian membrane-bound adenylyl cyclases. It is active as homodimer. Therefore, the structure determination of Rv1625c is important and urgent for exploring the structure, the function and the regulation of the membrane-bound adenylyl cyclases, and for answering the questions about the involvement of adenylyl cyclases in the pathogenesis of M. tuberculosis.
Structure and function of human NK cell receptors
Stuctural analysis of the NKC encoded, C-type lectin like receptor-ligand pair Nkp80 and AICL
Natural killer cells (NK cells) are lymphocytes of the innate immune system that play an important role in immune responses against certain pathogen-infected cells and tumors. Activity of NK cells is tightly regulated by the balance of activating and inhibitory signals delivered through a diverse set of cell surface receptors. Most of the NK receptors are members of the immunglobulin superfamily (IgSF) or the C-type lectin superfamily (CLSF) with the latter ones mostly being homo- or heterodimers of type II transmembrane proteins containing a single extracellular C-type lectin-like domain (CTLD). The CTLD was originally identified in certain lectins where it serves as a Ca2+-dependent carbohydrate recognition domain. However, most NKC-encoded CTLR have lost critical CTLD residues for carbohydrate binding and interact with protein ligands instead.
Although the CTLD-fold is a very common motive among proteins involved in the innate immune response, CTLDs from different proteins show only little sequence similarity (~20% to 40%) and have no conserved binding surface for dimerization or their ligand binding epitopes underlining the need for continued structure determination, even within a family of homologous protein modules. The crosstalk between the NK-cell receptor NKp80 and its ligand, the activation induced C-type lectin (AICL), may play a crucial role in immunosurveillance of viral and bacterial infections, malignancy, and the pathogenesis of autoimmune diseases, but the signal-transducing elements associated with NKp80 or AICL have not yet been defined. In addition, it is still unclear, whether NKp80 has more than one ligand or functions as coreceptor, e.g., with NKp46. Furthermore, only very little is known about AICL and its molecular binding properties to NKp80. Therefore, knowledge about the 3D-structure of both AICL and NKp80 as well as of their complex may help to get answers to the many questions left in the contect of NKp80-mediated NK-cells response and the role of its newly discovered ligand AICL.
Structural basis of ligand-mediated signalling in eukaryotes and plants
Deciphering the structure of Insulin Receptor Substrate
Insulin Receptor Substrate-1 (IRS-1) is a key element in Insulin and Insulin-like Growth Factor (IGF) actions. It transduces pleiotropic effects on cellular functions and modulates the processes such as metabolism, growth, cell differentiation and survival. Human IRS-1 is a ~170 KDa phospho-protein and is composed of 1242 amino acids. It contains Pleckstrin-Homology (PH) and Phospho-tyrosine binding (PTB) domains at the N-terminus that are involved in its targeting and binding to receptor tyrosine kinase(s). The remaining part of IRS1 (~980 amino acids) are collectively referred to as the "C-terminal tail" of IRS-1. The "C-terminal tail" region has various physiologically relevant motifs that are the targets of multiple kinases and other regulatory proteins. We propose that the "C-terminal tail" is a multi-domain region that contains numerous functional domains for protein-protein interaction and intracellular signal transduction. The aim of this project is to elucidate the structure of the "C-terminal tail" region of the IRS-1. This will ultimately help us to highlight the structural basis of the IRS-1 function.