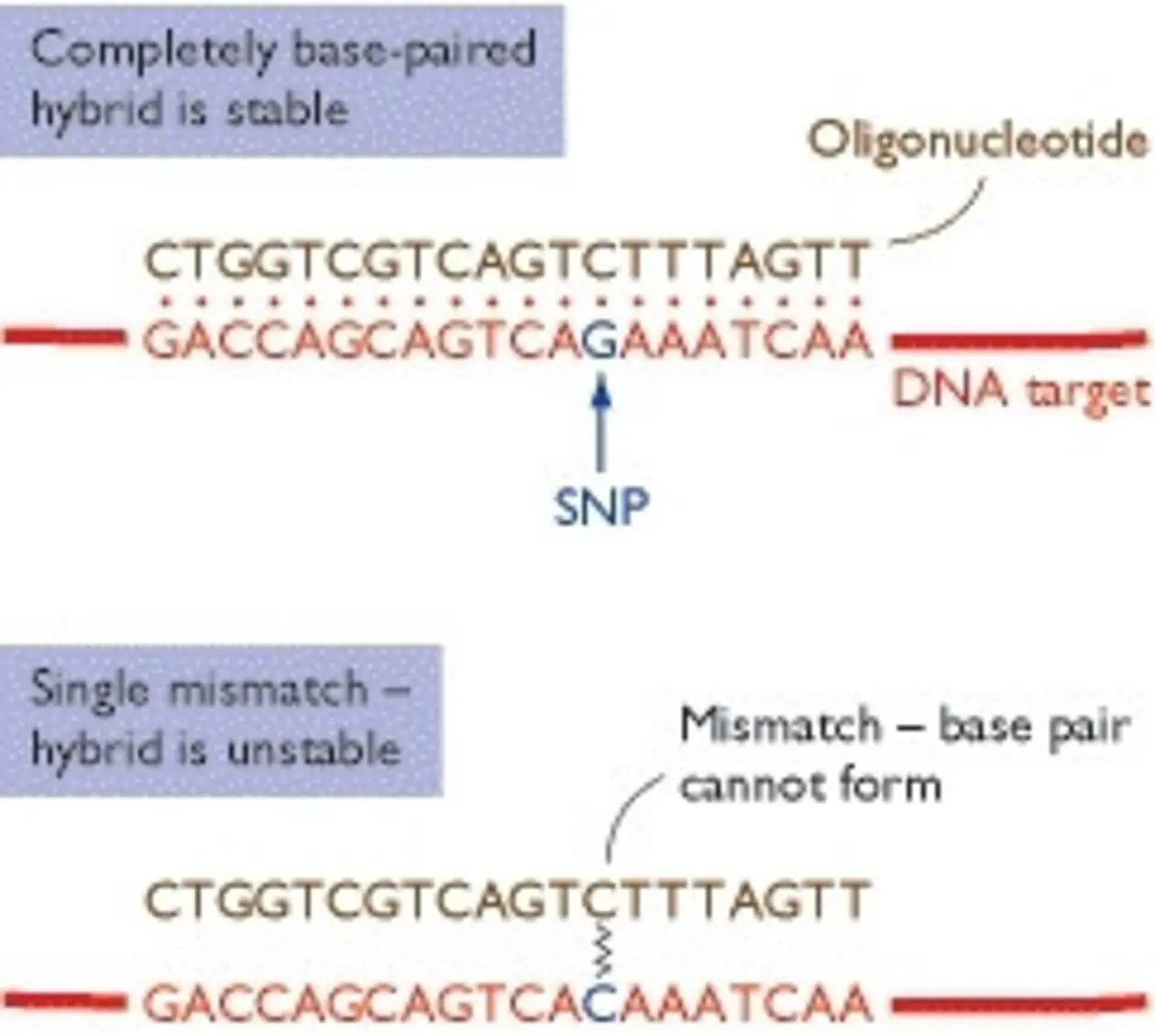
In contrast to other “gene-centric” microarray approaches, which are typically designed to be complementary to known (annotated) genes or expressed sequence tags (ESTs), tiling arrays interrogate a whole genome or large genomic region with probe features tiling the sequence of interest with a regular spacing. Due to their design, they are a very versatile experimental tool for studying an organism’s genome or transcriptome in a manner that is not biased by the current state of its genome annotation. Beside the main application of studying genome transcription, tiling arrays have been applied to e.g. discover new genes, analyze alternative splicing.
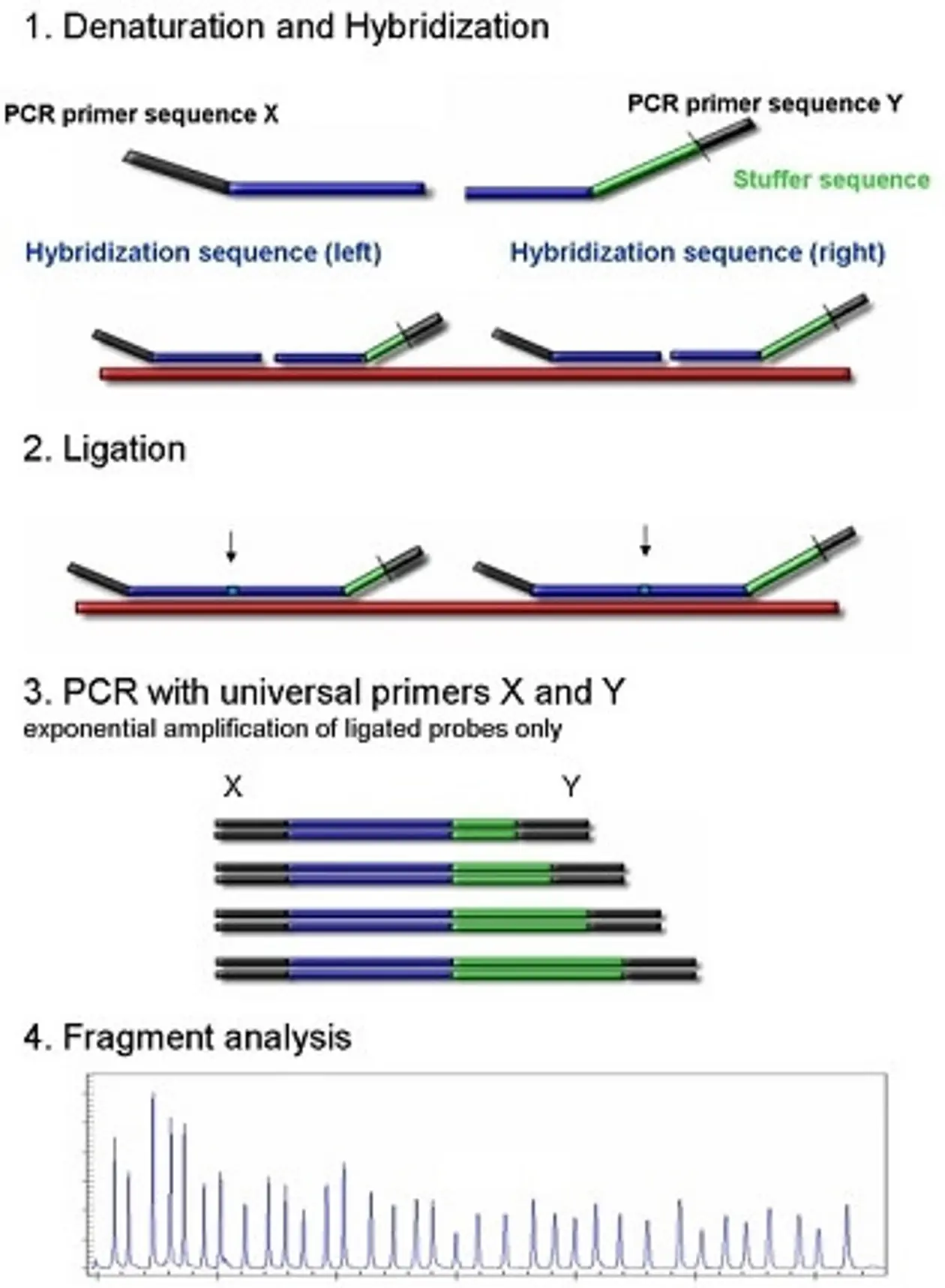
Analog to the microarray approach described above, tiling arrays consist of short fragment probes (25-1000 bp) specifically designed to cover contiguous regions of or even the entire genome, which have been immobilized at a specific position and quantity on a solid surface. Depending on probe lengths and spacing different degrees of resolution can be achieved.
Commonly, RNA is used as a template, which is converted into cDNA using reverse transcriptase, (fluorescent-)labelled and hybridized to the arrays (see above “Whole genome scan by SNP arrays”). After washing away the non-targeted cDNA the fluorescence signal intensity represents transcription level of hybridized templates.
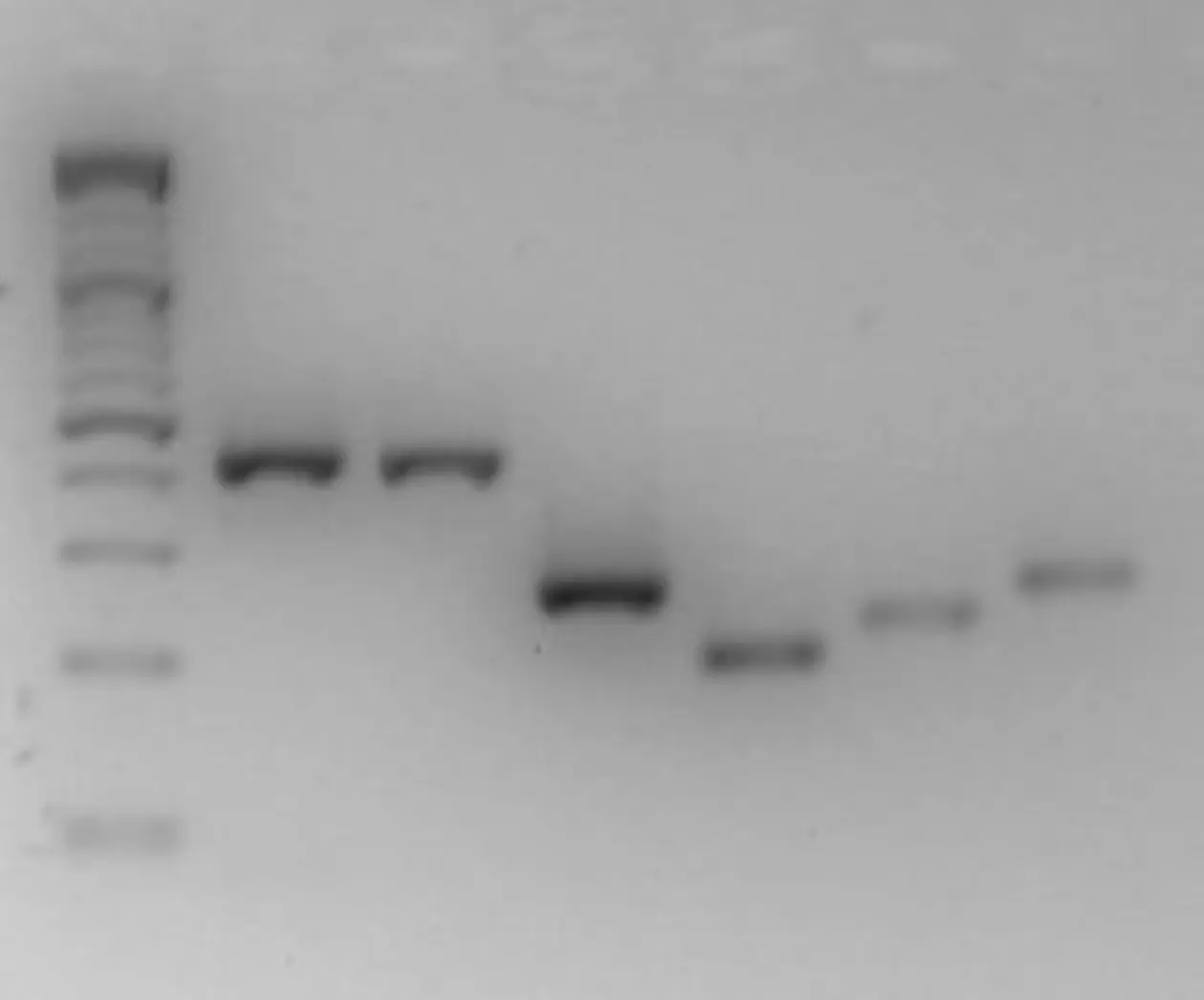
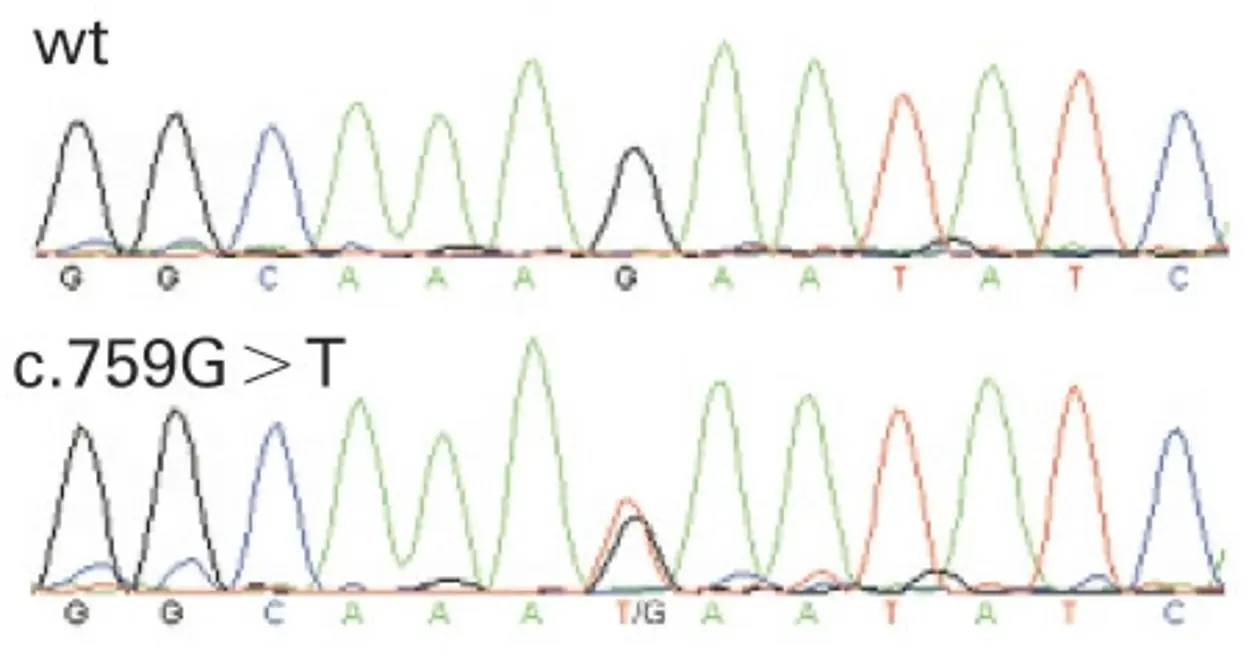
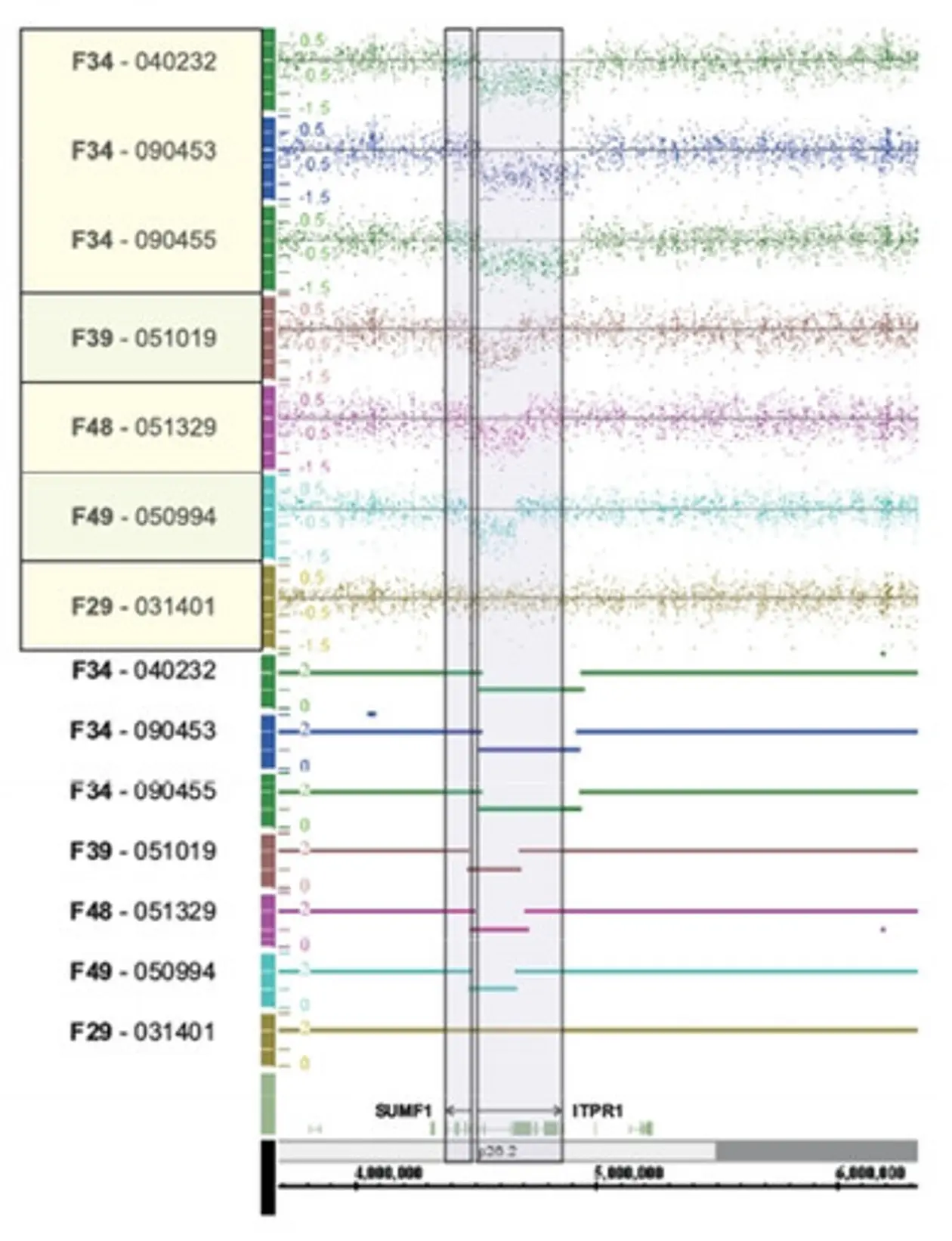
Alternatively, tiling arrays may be used to screen for macrodeletions. In this case templates of genomic DNA are hybridized to the array and the intensity of the signal indicates copy numbers of the respective DNA fragment (comparative genomic hybridization – CGH).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}